该文档可以帮助读者入门 Linux 系统,掌握常用命令和多种软件安装方式。此外还简述了全基因组测序技术,希望读者可以通过此文档初步了解生物信息学。
# Linux 初识
# 目录结构与路径
# 根目录
根目录(root)是系统最顶级目录,以 “/” 表示,其他目录均为根目录的下级目录。
# 家目录
家目录(home)是用户的最顶级目录,以 “~/” 表示,用户所有的目录均为家目录的下级目录。
# 绝对路径
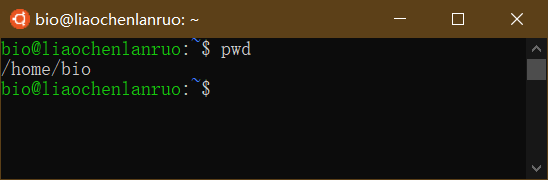
绝对路径指的是目录或文件的绝对位置,是从根目录开始的完整路径,如 “/home/bio/”。可通过 “pwd” 命令获取当前目录的绝对路径。
# 相对路径
相对路径不需要从根目录开始,只要指定与当前目录的相对位置即可。
# 当前目录
当前所在的路径,以 “./“表示。
# 上级目录
也称为父目录,以 “../“表示向上一级的目录,以 “../../” 表示向上两级的目录,以此类推。
# 命名法则
- 文档与目录均以英文命名,可使用字母、数字和下划线;
- 文档与目录的名称不允许存在空格;
- 名称区分大小写。
# 终端工具
终端(terminal)是运行 Linux 命令的工具,类似于 Windows 的命令行工具。Linux 各发行版均自带终端。

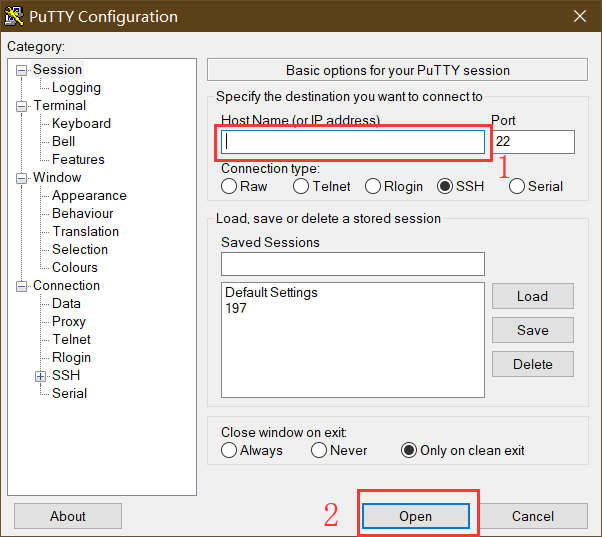
远程操控 Linux 服务器时,可以使用第三方的终端工具,如 PuTTy 软件。输入 IP 地址即可远程登录服务器运行命令。
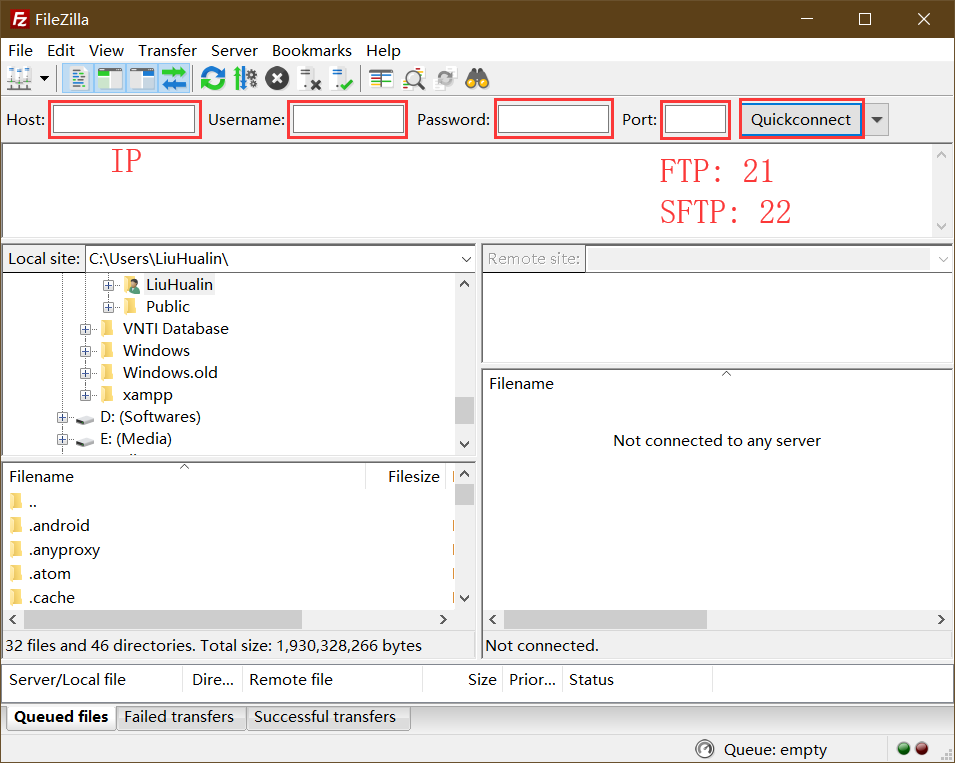
本地计算机与服务器之间的文件传输可以通过 FTP 软件实现,如 FileZilla。输入服务器的 IP 地址、用户名、密码以及端口即可链接服务器。如果服务器采用的时 FTP 协议,则端口填写 21,若采用的是 SFTP 协议,则端口设置为 22。
# 常用的命令
pwd: 获取当前位置的绝对路径
$ pwd |
mkdir: 创建目录
$ mkdir |
ls: 查看当前目录包含的内容
$ ls |
查看所有的目录和文件(包含隐藏的内容)
$ ls -a |
查看根目录所含内容
$ ls / |
查看家目录所含内容
$ ls ~/ |
cd: 切换路径
进入 “tools” 目录
$ cd tools |
vim: 创建 / 编辑文档
以下所有操作均需在英文输入法状态下进行。首先创建一个新文档 “example.txt”,并输入内容。
$ vim example.txt |

此时无法输入内容,需要按一下字母 “i” 键切换到输入模式。当左下角出现 “--NSERT --” 字样时,可以输入文字。

输入相关的内容。
输入完毕时需要先按一下 “ESC” 键退出编辑模式,此时 “--NSERT --” 字样消失。
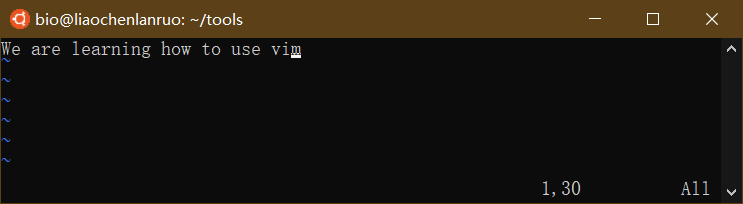
按住组合键 “shift + :” 切换到 vim 操作模式,此时左下角出现一个 “:”。
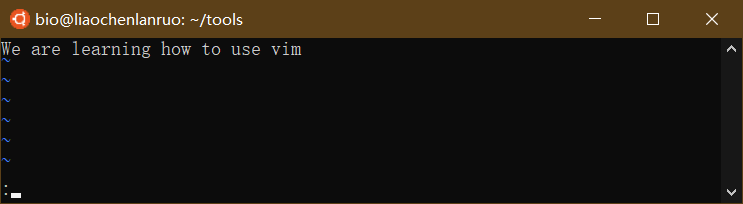
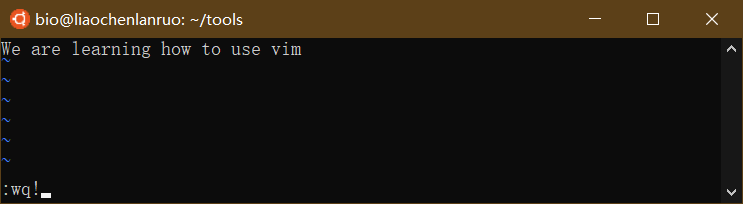
输入 “wq!” 保存修改并退出。
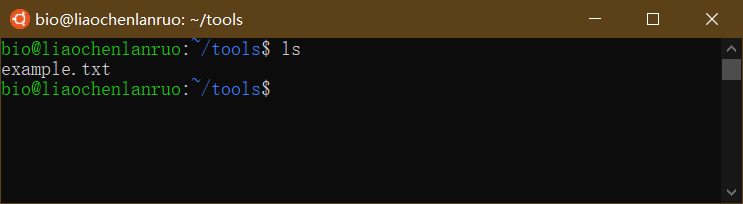
查看创建的文件是否在于目录下。
$ ls |
cp: 复制目录或文件
将创建的 “example.txt” 文档复制到上一级目录下。
$ cp example.txt ../ |
查看上一级目录下是否存在刚刚复制的文档。
$ ls ../ |
rm: 删除目录或文件
删除 tools 目录下的 “example.txt” 文档。
$ rm example.txt |
查看文档是否被删除。
$ ls |
mv: 移动 / 重命名
将上一级目录下的 “example.txt” 文档移动到当前目录下。
$ mv ../example.txt ./ |
查看文档是否移动成功。
$ ls ../ | |
$ ls |
将 “example.txt” 文档重命名为 “examp2.txt”。
$ mv example.txt examp2.txt |
查看重命名结果。
wget: 下载
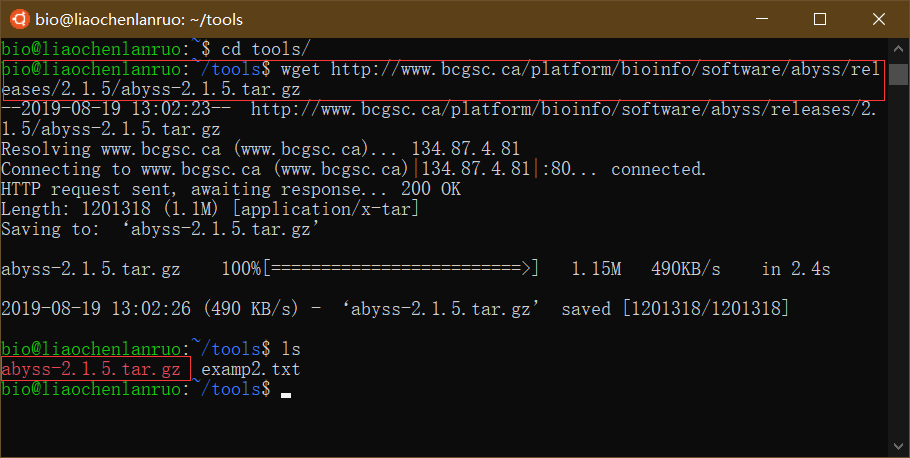
使用 wget 工具下载基因组拼接软件 “AbySS” 到 tools 目录下。
$ wget http://www.bcgsc.ca/platform/bioinfo/software/abyss/releases/2.1.5/abyss-2.1.5.tar.gz |
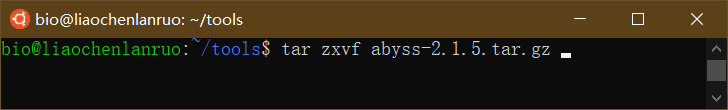
tar: 压缩 / 解压缩
tar.gz 格式的文件可用 “tar zxvf” 进行解压,将刚才下载的 “abyss-2.1.5.tar.gz” 解压缩。
$ tar zxvf abyss-2.1.5.tar.gz |
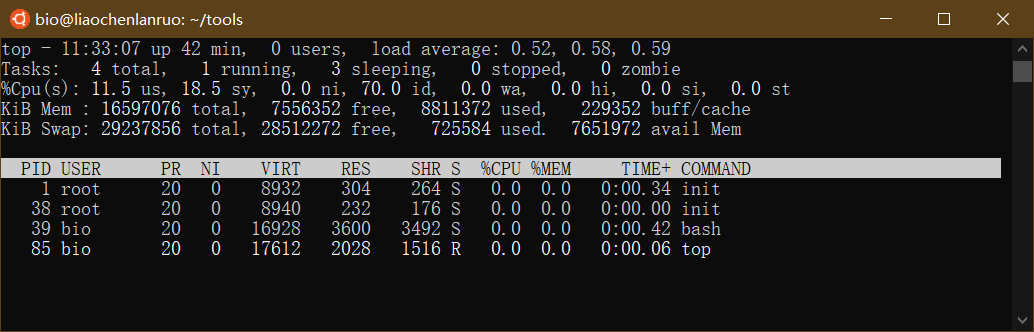
top: 查看系统进程
$ top |
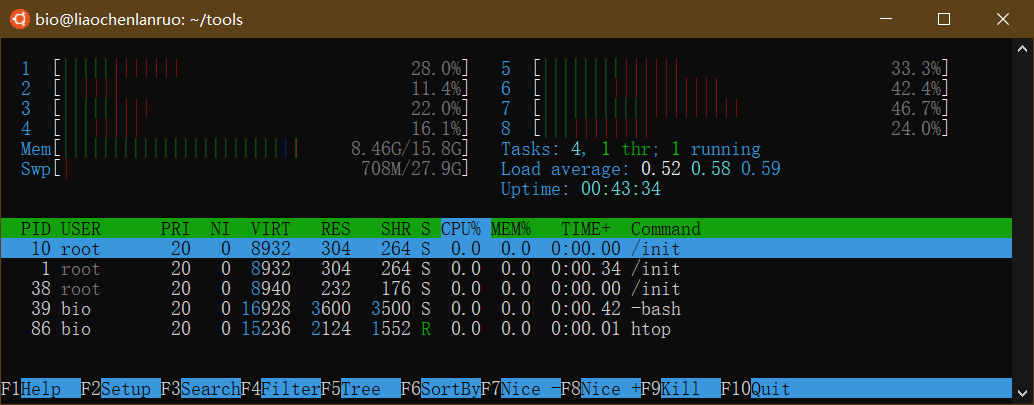
按字母键 “q” 退出。Ubuntu 还带有另一个更加直观的查看系统进程的工具 “htop”。
$ htop |
# 环境变量
在软件安装的时候经常需要设置环境变量,所谓的环境变量就是告诉计算机软件的安装位置。存放环境变量的文件在用户的家目录下,为隐藏文件,可通过 “ls -a” 命令查看。
$ ls -a ~/ |
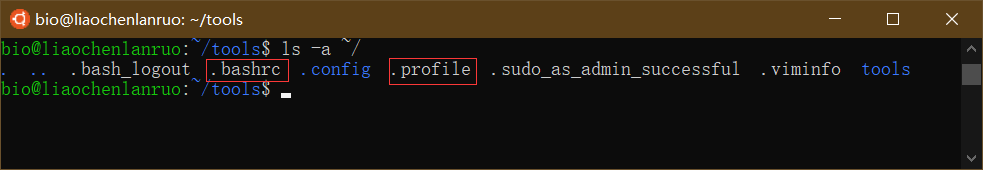
“.bashrc” 和 “.profile” 均为环境变量配置文件,通常我们只需要编辑 “.bashrc”。
# 软件安装
# 源码编译安装
源码安装适合于所有的 Linux 发行版以及 macOS。以刚下载的 “AbySS” 基因组拼接软件为例演示源码编译安装,一共分三步:配置(./configure)、编译(make)和安装(sudo make install)。首先进入 “AbySS” 软件目录下,并查看目录中的文件,找到配置文件 “configure”,根据 “README.md” 中的指示对软件进行配置。
$ cd abyss-2.1.5/ | |
$ ls |
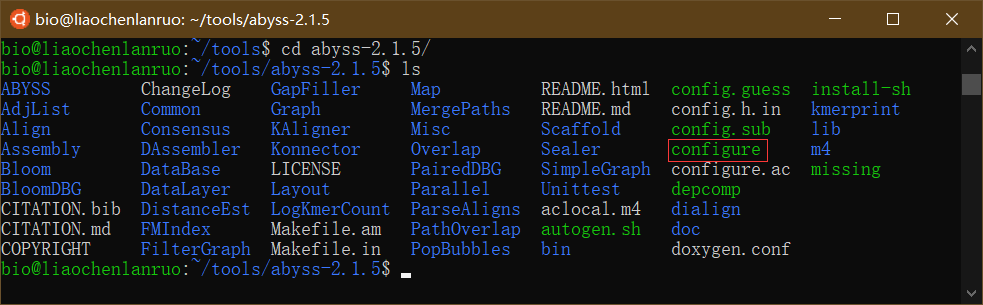
“./configure” 表示运行 configure 进行安装前配置。
$ ./configure |
进行编译
$ make |
进行安装,需要 “sudo” 命令提供对系统目录的写入权限。
$ sudo make install |
注意: 以上只演示了一般的安装方法,但是 “AbySS” 软件依赖其他的一些软件,需要先安装依赖包,最后安装 “AbySS”,否则会安装失败。
# 通过包管理工具安装
不同的 Linux 发行版具有各自的软件包管理器。目前常用的 Linux 发行版主要是基于 “RedHat” 和 “Debian” 而制作的。
RedHat 系列的包管理器为 “yum”,使用方法为在终端输入 “sudo yum install -y 软件名称”。
Debian 系列的包管理器为 “apt-get”,使用方法为在终端输入 “sudo apt-get install 软件名称”。
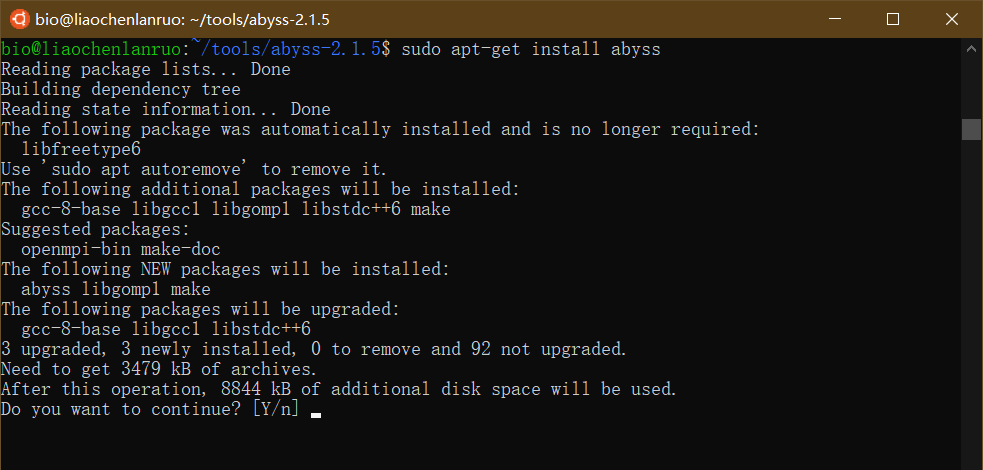
示例:通过 apt-get 在 Ubuntu 中安装 AbySS 软件,输入命令和密码后,根据提示输入 “Y” 并按回车键进行自动安装。
$ sudo apt-get install abyss |
# 添加环境变量
以原核生物基因预测软件 “Prodigal” 为例演示。首先在 Github 上找到 prodigal 的源码,点击 “Clone or download”,并按照图示点击链接右侧的图标以复制 git 地址。
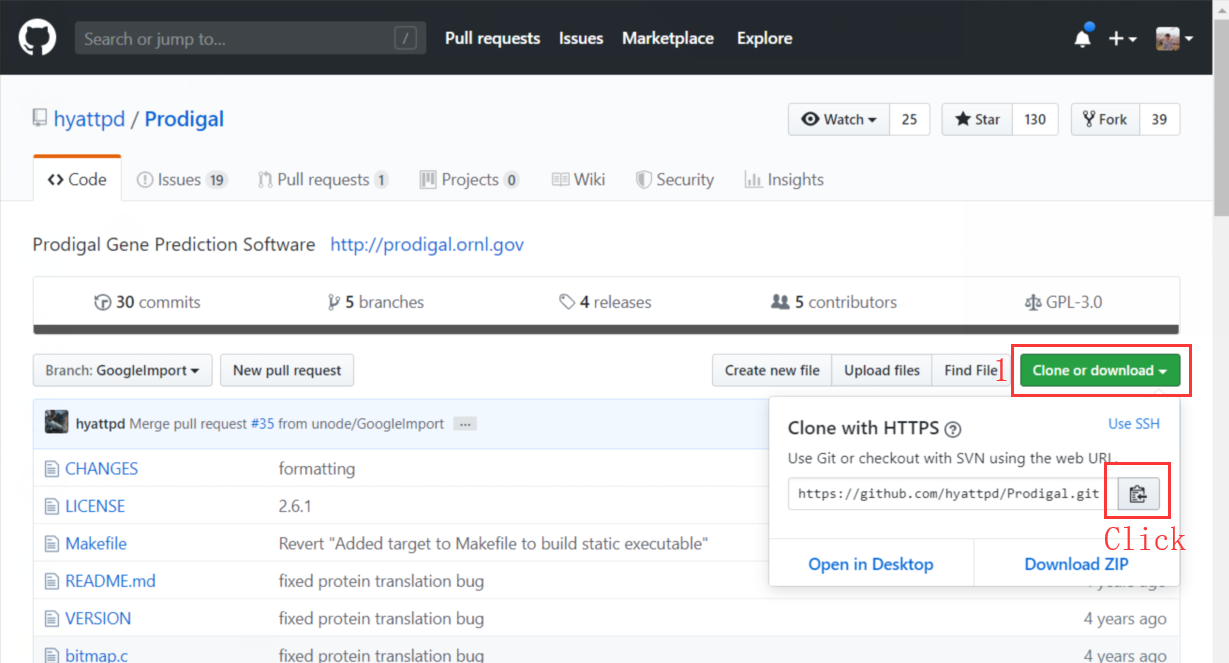
在终端中进入 tools 目录,并输入克隆命令将项目克隆到本地计算机。命令公式为 “git clone link”。
$ git clone https://github.com/hyattpd/Prodigal.git |
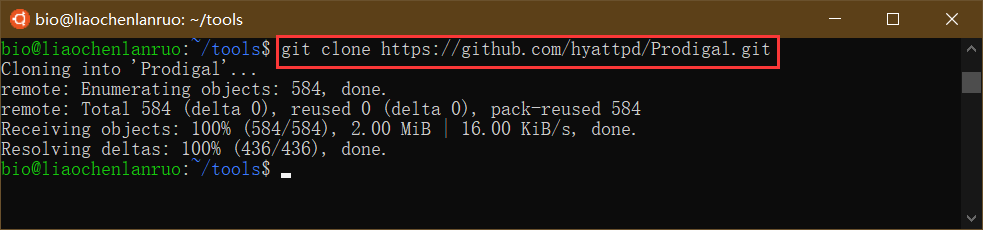
克隆完成后进入 “Prodigal” 目录。
$ cd Prodigal |
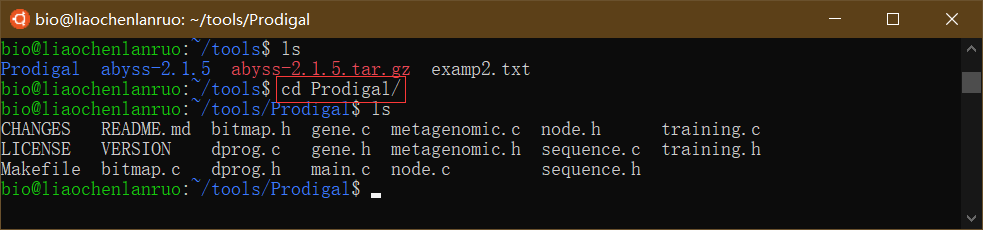
编译软件
$ make |
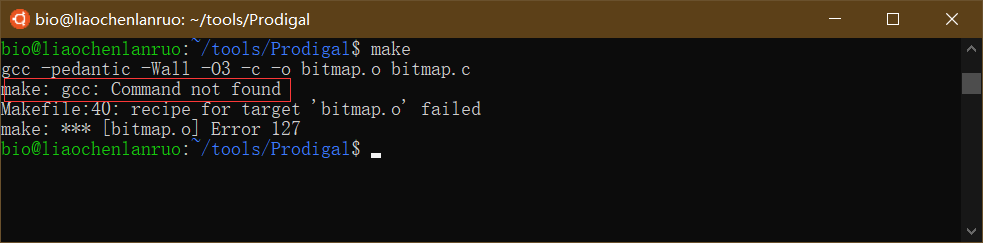
报错信息提示找不到 gcc 命令,因此需要首先安装 gcc,输入命令后根据提示输入密码,直至安装完成。
$ sudo apt-get install gcc |
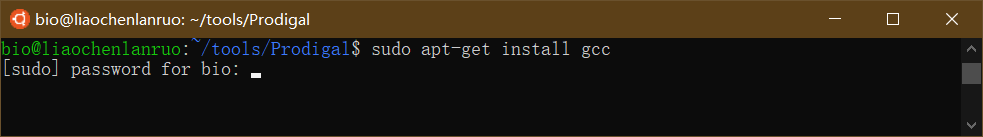
重新编译 prodigal
$ make | |
$ ls |
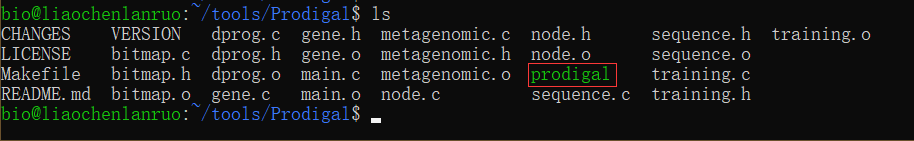
编译完成后得到了可执行程序,但是系统无法找到 prodigal 的路径,因此需要我们将其所在的路径加入到环境变量中。通过 vim 打开环境变量配置文件 “.bashrc”,进入编辑模式。
$ vim ~/.bashrc |
在文档末尾添加配置语句 “export PATH=HOME/tools/Prodigal” 。$HOME 代表家目录,“$HOME/tools/Prodigal” 代表 prodigal 可执行程序所在的目录。
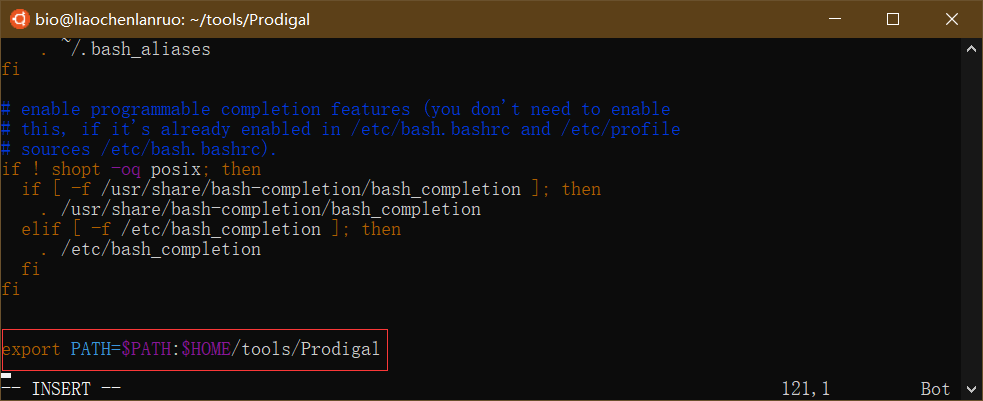
编辑完成后保存并退出。然后执行 “source ~/.bashrc” 命令刷新,通知系统 “.bashrc” 文档已经更改。
$ source ~/.bashrc |

测试配置是否成功。
$ prodigal -h |
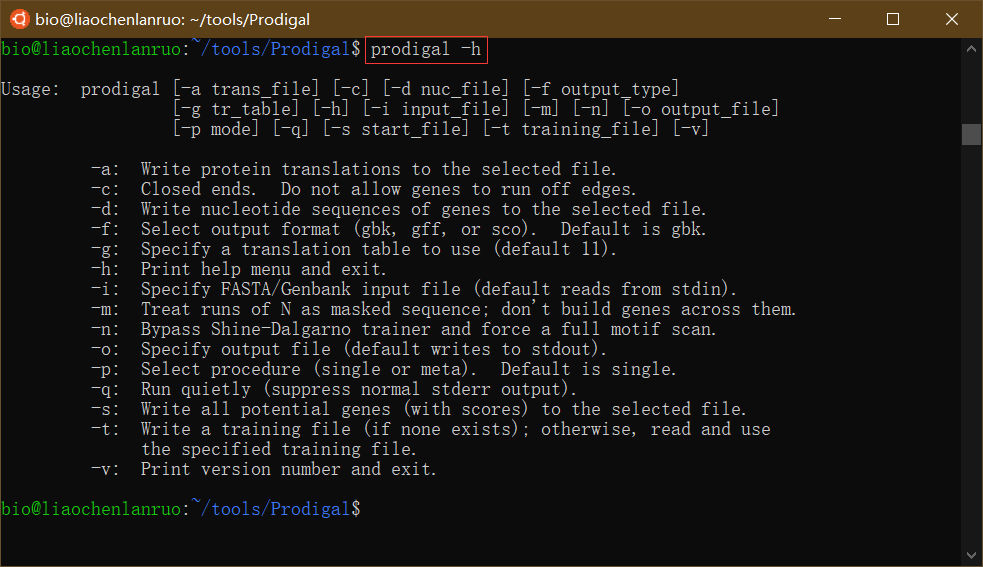
若要将其他软件加入到环境变量,只需在后面加入其他软件所在路径即可,各软件的路径间以英文 “:“分割,不得有空格。下图为将多个软件加入到环境变量的示例。

# 创建软链接
软链接(Soft Link)相当于 Windows 系统中的快捷方式,可以将可执行程序的软链接存放至系统默认的环境变量之中,如 “/usr/bin/” 或 “/usr/local/bin” 之中。仍旧以刚编译好的 prodigal 软件为例,创建软链接的公式为 “sudo ln -s /home/bio/tools/Prodigal/prodigal /usr/local/bin/prodigal” ,根据提示输入密码完成创建。
$ sudo ln -s /home/bio/tools/Prodigal/prodigal /usr/local/bin/prodigal |
通过 “whereis” 命令查看软链接是否创建成功。
$ whereis prodigal |
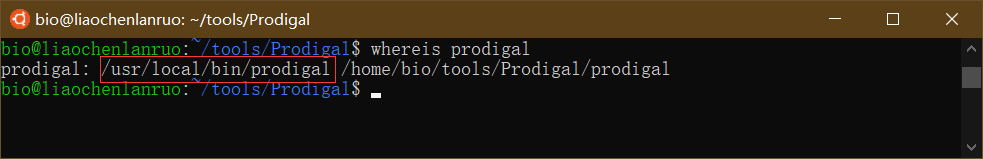
注意: 创建软链接时要输入绝对路径,否则会报错 “Too many levels of symbolic links”。
# 通过 Anaconda 包管理器进行安装
Anaconda 是一款比较易用的跨平台软件包管理器,Bioconda 是 conda 的一个通道,专门管理生物信息学软件。通过 conda 安装软件时可以一键安装所有的依赖包,大大节约了时间并降低了安装难度。Bioconda 目前有超过 600 个贡献者和 500 个成员,大部分生物信息学软件都被包含其中。用户可以到其官网搜索需要的软件是否被囊括其中。
(1)安装 conda
此处,我们安装 Miniconda,进入官网,选择适应自身系统及 python 版本的安装文件。
查看系统 python 版本
$ python -v |
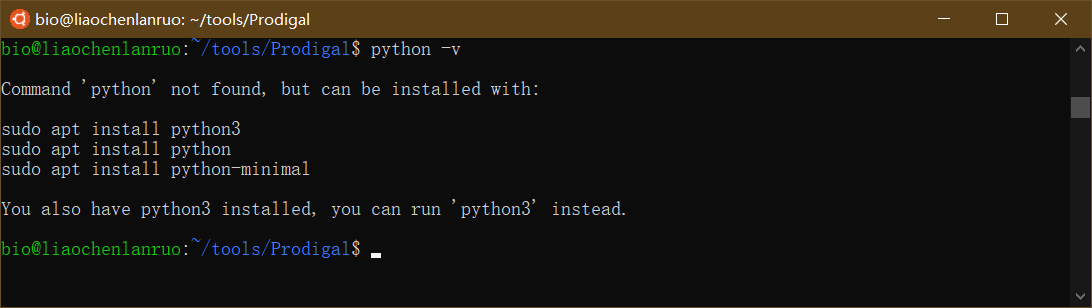
可以看出该系统已经安装了 python3,因此下载 Linux Python 3.7 64-bit (bash installer)。右键单击相应安装包获取链接,使用 wget 下载至 tools 目录下。建议用户安装 Python 3,因为 Python 软件基金会将于 2020 年元旦停止对 Python 2 的维护(https://pythonclock.org/)。
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh |
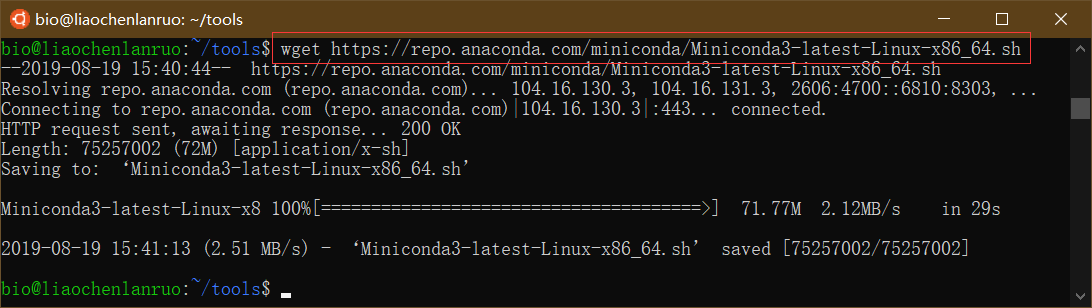
开始安装 Miniconda
$ bash Miniconda3-latest-Linux-x86_64.sh |
根据提示按 “Enter” 键查看 license,并输入 “yes” 按 “Enter” 继续,按 “Enter” 确认安装位置,miniconda 被安装到家目录下的 miniconda3 目录中。最后输入 “yes”,按 “Enter” 进行初始化。最后,通过 “source ~/.bashrc” 命令刷新。
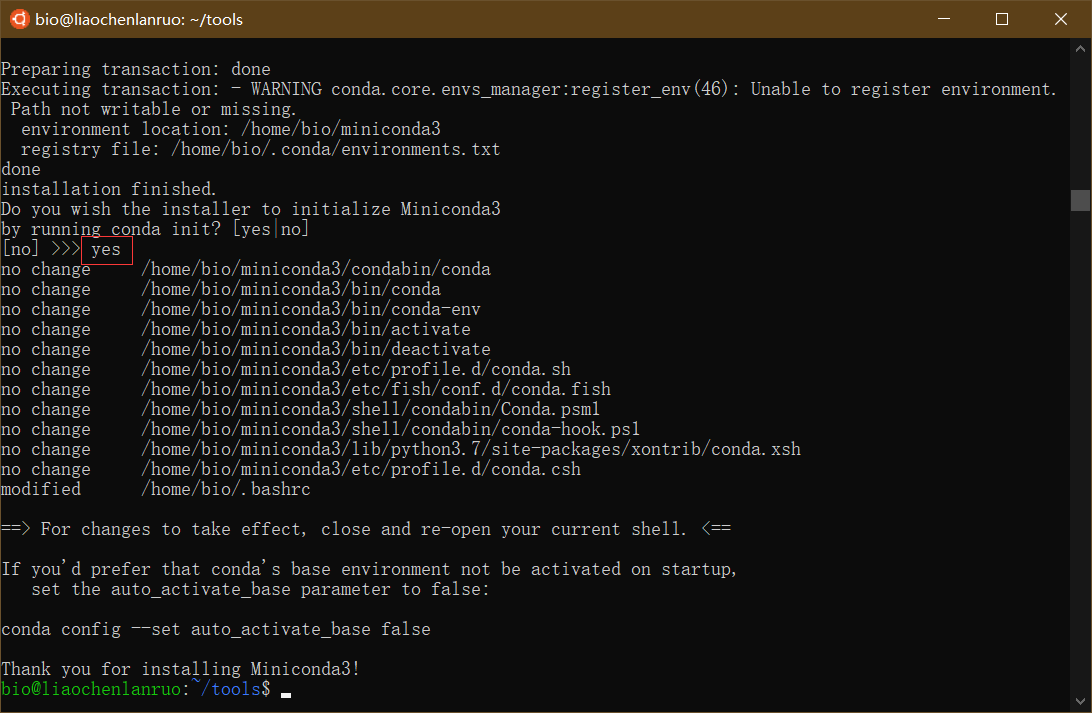
(2)设置 bioconda channel
在终端中输入以下三条命令添加 channels:
$ conda config --add channels defaults | |
$ conda config --add channels bioconda | |
$ conda config --add channels conda-forge |
至此,bioconda 配置完毕,可以通过 conda 安装生物信息学软件。下面通过 conda 安装 mapping 软件 “bwa”。
$ conda install bwa |
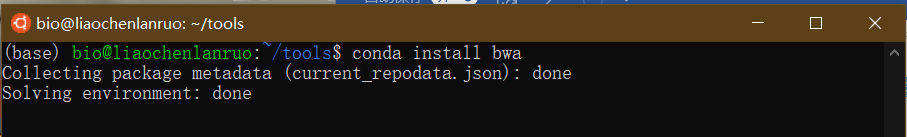
根据提示输入 “y” 完成安装。
# MacOS 相关操作
MacOS 与 Linux 系统相似,基本命令相同,但是软件安装存在一些差异。
# MacOS 安装生物信息学软件
# 源码安装
源码安装方式与 Linux 安装方式一致。
# 创建软链接
配置方法与 Linux 一致。
# 环境变量
MacOS 环境变量配置方法与 Linux 配置方法一致,但配置文件为家目录下的 “.bash_profile”,即运行如下命令进行编辑。
$ vim ~/.bash_profile |
编辑完成并保存后需要运行 source 命令。
$ source ~/.bash_profile |
# 包管理器
MacOS 的软件包管理器为 Homebrew,可以在终端中通过以下命令安装 Homebrew。
$ ruby -e "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/master/install)" |
通过 Homebrew 安装 mapping 软件 “bwa”。
$ brew install bwa |
# MacOS 配置 Anaconda
安装 Miniconda
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-MacOSX-x86_64.sh | |
$ sh Miniconda3-latest-MacOSX-x86_64.sh |
添加 Bioconda 通道
$ conda config --add channels defaults | |
$ conda config --add channels bioconda | |
$ conda config --add channels conda-forge |
安装软件 bwa
$ conda install bwa |
# 现代测序技术
# 二代测序 ("Next-generation" sequencing technology)
第二代测序技术的核心思想是边合成边测序(Sequencing by Synthesis),即通过捕捉新合成的末端的标记来确定 DNA 的序列。应用最广的技术平台主要为 Illumina 公司的产品。其优点为高通量、错误率低、成本低等。
- Illumina 测序中的几个名词
- Read length: 测序的 DNA 片段的碱基长度。
- Insert size: 双端测序时接头(adapter)中间序列的长度。
- Junction: insert 序列中间未被测序的部分。
- Flowcell: 是指 Illumina 测序时,测序反应发生的位置,1 个 flowcell 含有 8 条 lane。
- Lane: 每一个 flowcell 上都有 8 条泳道,用于测序反应,可以添加试剂,洗脱等等。
- Raw data: 测序完成后未去接头、引物以及去除低质量序列的数据。
- Clean data: 去除 Raw data 中的接头序列、linker、低质量 reads、长度较短的 reads 及核糖体 RNA 和 ncRNA 产生的 reads。
- 数据量: read 长度乘以 reads 数目。
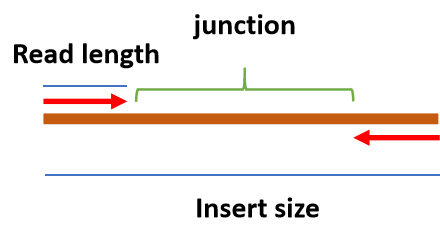
# 三代测序
三代测序又称为单分子测序,测序过程无需进行 PCR 扩增,可以产生超长的 reads,因此能够跨越高 GC 含量区域和高度重复区域。目前常用的测序平台包括 Pacific Biosciences(PacBio)和 Oxford Nanopore。
PacBio 以 SMRT Cell 为载体进行测序反应,SMRT Cell 是一张带有纳米孔的超薄金属片。PacBio 采用边合成便测序的方式,测序反应在纳米孔中进行,一个纳米孔中固定一个 DNA 聚合酶和一条 DNA 模板。延伸反应的过程中检测 dNTP 荧光信号以确定碱基顺序。
Oxford 开发的纳米单分子测序技术属于真正的实时测序,它基于电信号来判读碱基。
| Platform\Instrument | Throughput (Gb) | Read length (bp) | Strength | Weakness |
|---|---|---|---|---|
| Sanger sequencing | ||||
| ABI 3500/3730 | 0.0003 | Up to 1 kb | Read accuracy and length | Cost and throughput |
| Illumina | ||||
| MiniSeq | 1.7–7.5 | 1×75 to ×150 | Low initial investment | Run and read length |
| MiSeq | 0.3–15 | 1×36 to 2×300 | Read length, scalability | Run length |
| NextSeq | 10–120 | 1×75 to 2×150 | Throughput | Run and read length |
| HiSeq (2500) | 10–1000 | ×50 to ×250 | Read accuracy, throughput | High initial investment, run |
| NovaSeq 5000/6000 | 2000–6000 | 2×50 to ×150 | Read accuracy, throughput | High initial investment, run |
| IonTorrent | ||||
| PGM | 0.08–2 | Up to 400 | Read length, speed | Throughput, homopolymers |
| S5 | 0.6–15 | Up to 400 | Read length, speed | Homopolymers |
| Proton | 10–15 | Up to 200 | Speed, throughput | Homopolymers |
| Pacific BioSciences | ||||
| PacBio RSII | 0.5–1 | Up to 60 kb | ead length, speed (Average 10 kb, N50 20 kb) | High error rate and initial |
| Sequel | 5–10 | Up to 60 kb | Read length, speed (Average 10 kb, N50 20 kb) | High error rate |
| Oxford Nanopore | ||||
| MInION | 0.1–1 | Up to 100 kb | Read length, portability | High error rate, run length |
# 常见序列格式
# Fastq
我们得到的下机序列一般为 fastq 格式,每一条 read 包含 4 行,第一行为测序仪器信息以及测序信息,第二行为碱基序列,第三行一般无信息,第四行为对应第二行中每个碱基的测序质量信息。

| Strings | Description |
|---|---|
| @ST-E00310 | The unique instrument name |
| 147 | The run id |
| HVT25CCXX | The flowcell id |
| 3 | Flowcell lane |
| 1011 | The number within the flowcell lane |
| 13382 | ‘x’-coordinate of the cluster within the title |
| 1819 | ‘y’-coordinate of the cluster within the title |
| 1 | The number of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| N | Y if the read fails filter (read is bad), N otherwise |
| 0 | 0 when none of the control bits are on, otherwise it is an even number |
| TGAAGACA | Index sequence |
# Fasta
FASTA 格式为文本文档,内含核苷酸或氨基酸序列以及其 IDs。每条序列包含两部分,第一部分为 ID 及注释信息,以 ">" 开头,at the start, 第二部分为核苷酸序列或氨基酸序列。

# Genbank
GenBank 格式包含了基因组序列和注释信息。

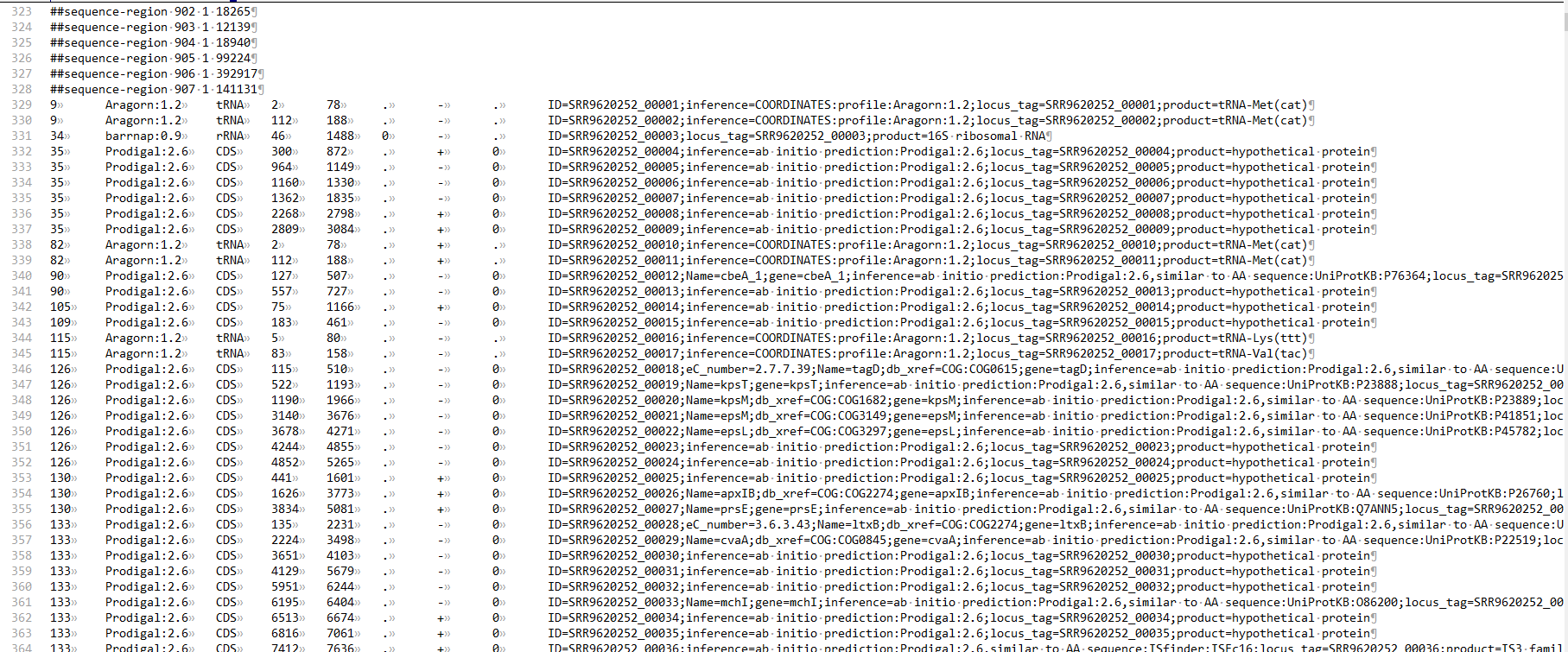
# GFF3
GFF3 (Generic Feature Format version 3) 格式描述了序列的特征,每一行含有 9 列数据,列与列之间以制表符分割。
# 基因组拼接基本概念
# 测序深度
测序深度(Sequencing depth)指测序得到的总碱基数(read 长度 x reads 数目)与待测基因组大小的比值。假设一个基因组大小为 2M,测序深度为 10X,那么获得的总数据量为 20M。
# 测序覆盖度
指测序获得的序列占整个基因组的比例。由于基因组中的高 GC、重复序列等复杂区域的存在,测序获得的序列经常无法覆盖基因组上所有的区域。例如覆盖度是 96%,表明还有 4% 的序列区域未测到。
# Read、Contig、Scaffold
测序得到的序列被称作 reads,在一个 read 中连续的 N 个碱基所组成的序列称作 k-mer,把 k-mer 集合拼接起来形成的长 DNA 序列称为 contig。通过 pair ends 信息将 contigs 按顺序进行排列得到 scaffold。
# N50
将 contigs 或 scaffolds 根据长度从大到小排列并累加,当其累计长度达到全部组装序列总长度的 50% 时,加上去的最后一个 contig 或 scaffold 的大小即为 N50 的大小,N50 是评价基因组拼接质量的重要参数。
# 参考文献
Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clinical Microbiology and Infection, 2018, 24: 335-341
