
承接上一篇文章 Swissprot 数据库的本地化与序列比对。
# 应用场景
分别预测了多个样本 / 基因组中某些基因的存在与否即数量,需要将这些样本 / 基因组中的基因数量情况合并在一起构建矩阵,此时,手动是非常困难和无趣的。又该请出 Perl 神了。
# 输入文件
在上一篇文章中,我们将多个宏基因组蛋白序列与 SwissProt 数据库做了比对,并根据比对到的 ID 与其他数据库做了 mapping,得到了多个输出文件,保存为 “sample.mapped”。其中 “sample” 可以是样本名,也可以是基因组名,它将出现在最后构建的矩阵中。这些文件既可作为本例的输入文件,其内容大概是下面酱紫的,各列以制表符分隔。
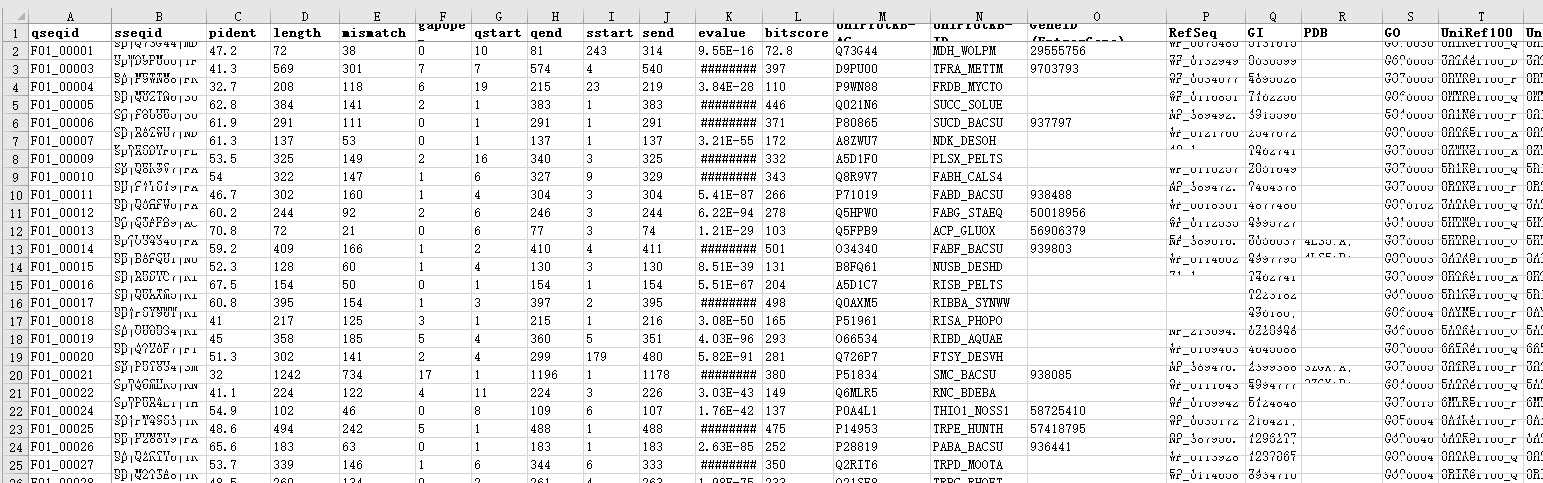
# 写脚本
上图是其中一个文件的内容的一部分,接下来我们将提取第 19 列的 GO number 来构建矩阵。将以下代码保存到文件中,命名为 “get_matrix.pl”。
#!/usr/bin/perl | |
use strict; | |
use warnings; | |
# Author: Liu hualin | |
# Date: Sep 29, 2021 | |
my %hash; | |
my %ids; | |
my %samples; | |
my @files = glob("*.mapped"); | |
foreach (@files) { | |
$_=~/(.+).mapped/; | |
my $sample = $1; | |
$samples{$1}++; | |
open IN, "$_" || die; | |
<IN>;# 忽略第一行,如果第一行不是标题行,请将该行注释掉 | |
while (<IN>) { | |
chomp; | |
my @lines = split /\t/; | |
if ($lines[18]=~/.+\; /) { | |
my @terms = split /\; /, $lines[18];# 18 代表文件的第 19 列,若想提取其他列,可以自行修改该数字为 “列号 - 1”,因为第一列代号为 0 | |
foreach (@terms) { | |
$ids{$_}++; | |
$hash{$sample}{$_}++; | |
} | |
}elsif ($lines[18]=~/\S+/) { | |
$ids{$lines[18]}++; | |
$hash{$sample}{$lines[18]}++; | |
} | |
} | |
} | |
open OUT, ">Matrix.txt" || die; | |
my @samples = sort keys %samples; | |
my @ids = sort keys %ids; | |
print OUT "\t" . join("\t", @samples) . "\n"; | |
for (my $i=0; $i<@ids ;$i++) { | |
print OUT $ids[$i]; | |
for (my $j=0; $j<@samples ;$j++) { | |
if (exists $hash{$samples[$j]}{$ids[$i]}) { | |
print OUT "\t$hash{$samples[$j]}{$ids[$i]}"; | |
}else { | |
print OUT "\t0"; | |
} | |
} | |
print OUT "\n"; | |
} | |
close OUT; |
# 构建矩阵
将脚本与输入文件放在同一目录下,在终端或 Windows 命令行中运行如下命令,得到的 “Matrix.txt” 即为输出文件。
perl get_matrix.pl |
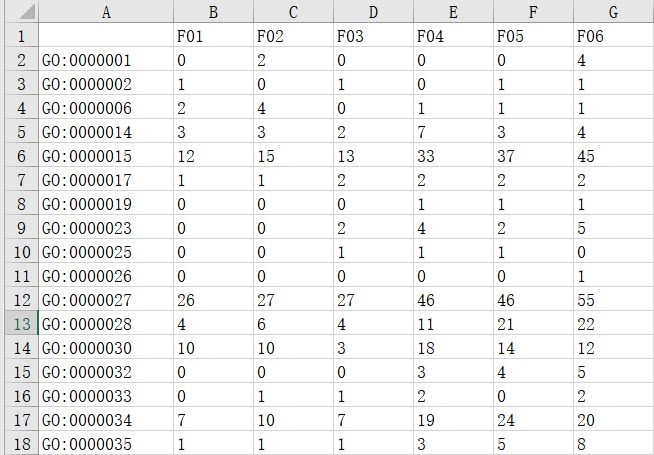
# 脚本获取
关注公众号 “生信之巅”,聊天窗口回复 “412a” 获取下载链接。
 |  |
敬告:使用文中脚本请引用本文网址,请尊重本人的劳动成果,谢谢!
