
# 数据库下载与构建
# 下载
wget https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz |
# 构建
解压缩
gunzip -d uniprot_sprot.fasta.gz
构建 blast + 数据库
makeblastdb -in uniprot_sprot.fasta -dbtype prot -out uniprot_sprot -parse_seqids
构建 DIAMOND 数据库
diamond makedb --in uniprot_sprot.fasta -d uniprot_sprot_diamond
# 比对
blastp 蛋白比对
blastp -query F01.faa -out F01.swissprot -db /new_data/hualin/db/uniprot_sprot -outfmt 6 -num_threads 30 -evalue 1e-5
diamond 蛋白比对
单个基因组对比
diamond blastp -d /new_data/hualin/db/uniprot_sprot_diamond -q F01.faa -e 1e-5 -f 6 -o F01.diamond -k 1 --sensitive -p 30 --query-cover 50
多个个基因组对比
不会 shell 没办法,写 Perl 脚本 (run_diamond.pl) 来完成。
#!/usr/bin/perluse strict;
use warnings;
# Author: Liu hualin# Date: Sep 28, 2021my @faa = glob("*.faa");# 读取所有后缀为 “.faa” 的文件,可以自己更改
foreach (@faa) {
$_=~/(.+).faa/;
my $out = $1 . ".diamond";
# 将 /new_data/hualin/db/uniprot_sprot_diamond 换成自己的数据库路径;-p 表示线程数,在笔记本上用 6 个即可system("diamond blastp -d /new_data/hualin/db/uniprot_sprot_diamond -q $_ -e 1e-5 -f 6 -o $out -k 1 --sensitive -p 30 --query-cover 50");
}将上述代码复制到文件中,命名为 “run_diamond.pl”,将其和序列文件放在同一目录下,并在终端中输入如下命令,完成分析:
perl run_diamond.pl
# 将比对结果 mapping 至其他数据库
打开网址 https://www.uniprot.org/uploadlists/, 上传比对上的 swissprot ID,可以将比对结果转换为诸如 KEGG 等其他数据库的 ID。个人感觉不是很好用。
我们可以把 mapping 文件下载下来,自己写脚本来提取信息,虽然麻烦些,但得到的更多。
下载 mapping 文件
wget https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/idmapping_selected.tab.gz解压缩
gunzip -d idmapping_selected.tab.gz
写脚本提取对应信息
Diamond 比对的结果文件内容如下,第一列是自己的氨基酸序列 ID,第二列是 SwissProt 数据库中序列的 ID,而我们真正需要的是第二列中两个竖线中间的内容,在稍后的脚本中将通过正则表达式把它给揪出来。
F01_00001 sp|Q73G44|MDH_WOLPM 47.2 72 38 0 10 81 243 314 9.55e-16 72.8
F01_00003 sp|D9PU00|TFRA_METTM 41.3 569 301 7 7 574 4 540 4.89e-131 397
F01_00004 sp|P9WN88|FRDB_MYCTO 32.7 208 118 6 19 215 23 219 3.84e-28 110
F01_00005 sp|Q021N6|SUCC_SOLUE 62.8 384 141 2 1 383 1 383 1.45e-155 446
开始写脚本,保存为 “run_mapping.pl”。
#!/usr/bin/perluse strict;
use warnings;
# Author: Liu hualin# Date: Sep 28, 2021my %maps;
my @diaout = glob("*.diamond");# 读取所有的 diamond 比对后的输出文件
foreach (@diaout) {
$_=~/(\S+).diamond/;
my %hash;
open IN, "$_" || die;
while (<IN>) {
chomp;$_=~s/[\r\n]+//g;
my @lines = split;
$lines[1]=~/.+\|(.+)\|.+/;
$hash{$1}++;
}close IN;open IN, "idmapping_selected.tab" || die;
while (<IN>) {
chomp;$_=~s/[\r\n]+//g;
my @lines = split;
if (exists $hash{$lines[0]}) {
$maps{$lines[0]} = $_;
}}close IN;}my @diaout2 = glob("*.diamond");# 读取所有的 diamond 比对后的输出文件
foreach (@diaout2) {
$_=~/(\S+).diamond/;
my $out = $1 . ".mapped";
open IN, "$_" || die;
open OUT, ">$out" || die;
print OUT "qseqid\tsseqid\tpident\tlength\tmismatch\tgapopen\tqstart\tqend\tsstart\tsend\tevalue\tbitscore\tUniProtKB-AC UniProtKB-ID GeneID (EntrezGene) RefSeq GI PDB GO UniRef100 UniRef90 UniRef50 UniParc PIR NCBI-taxon MIM UniGene PubMed EMBL EMBL-CDS Ensembl Ensembl_TRS Ensembl_PRO Additional PubMed\n";
while (<IN>) {
chomp;$_=~s/[\r\n]+//g;
my @lines = split;
$lines[1]=~/.+\|(.+)\|.+/;
if (exists $maps{$1}) {
print OUT $_ . "\t" . $maps{$1} . "\n";
}else {
print OUT $_ . "\n";
}}close IN;close OUT;}将脚本与 diamond 比对的结果文件以及下载的 mapping 文件放在同一目录下,在终端里输入如下命令即可得到 mapping 后的结果:
perl run_mapping.pl
# GO 注释
从 map 后的文件中提取基因 ID 和 GO number,各列以制表符分隔,没有 GO 注释的只输出 gene ID。
准备脚本,命名为 “get_GO.pl”,与上一步生成的 “*.mapped” 文件放在同一目录下。
#!/usr/bin/perluse strict;
use warnings;
# Author: Liu hualin# Date: Sep 28, 2021my @mapped = glob("*.mapped");
foreach (@mapped) {
$_=~/(.+).mapped/;
open IN, "$_" || die;
my $out = $1 . ".GO";
open OUT, ">$out" || die;
<IN>;
while (<IN>) {
chomp;$_=~s/[\r\n]+//g;
my @lines = split /\t/;
print OUT $lines[0];
if ($lines[18]=~/.+\; /) {
my @terms = split /\; /, $lines[18];# 18 代表文件的第 19 列
print OUT "\t" . join("\t", @terms) . "\n";
}elsif ($lines[18]=~/\S+/) {
print OUT "\t" . $lines[18] . "\n";
}else {
print OUT "\n";
}}close IN;close OUT;}在终端或者 Windows 命令行中运行如下命令,得到的 “*.GO” 为输出文件。
perl get_GO.pl
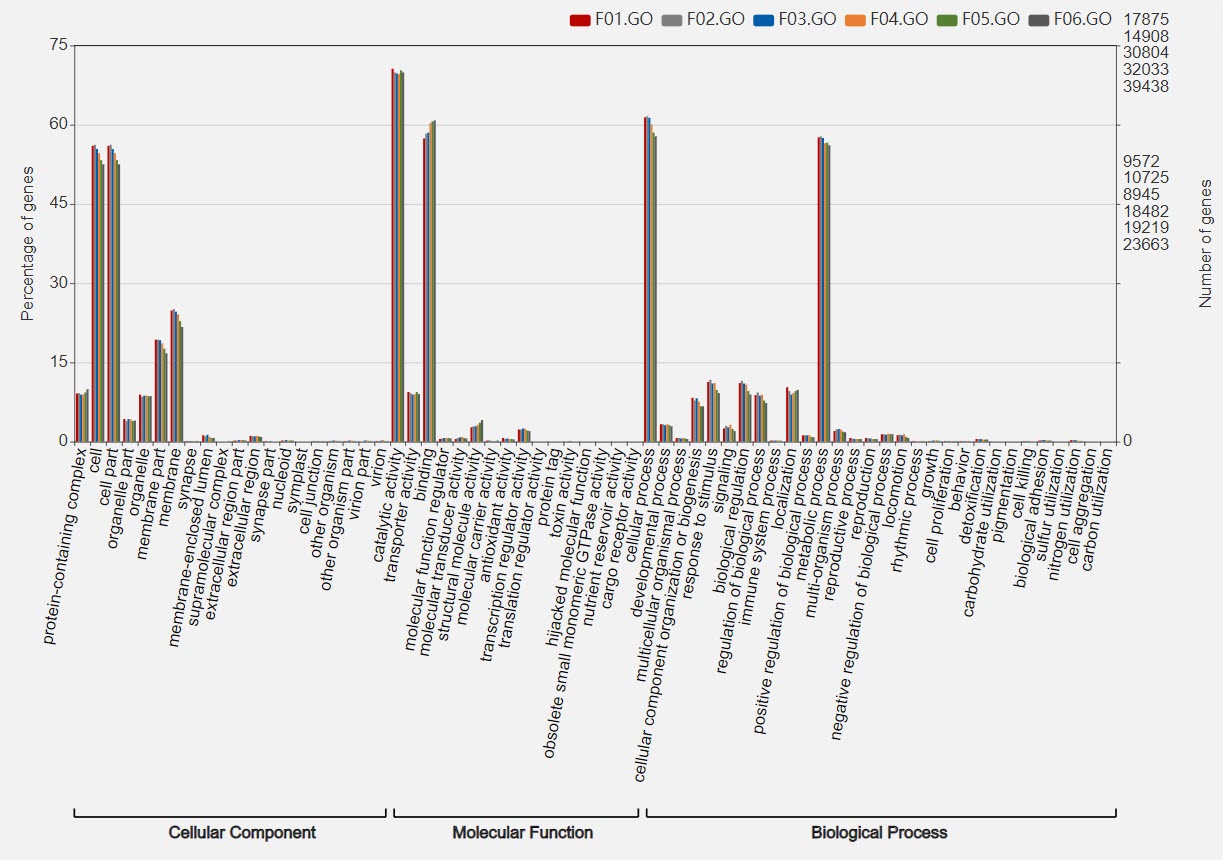
GO 注释与可视化
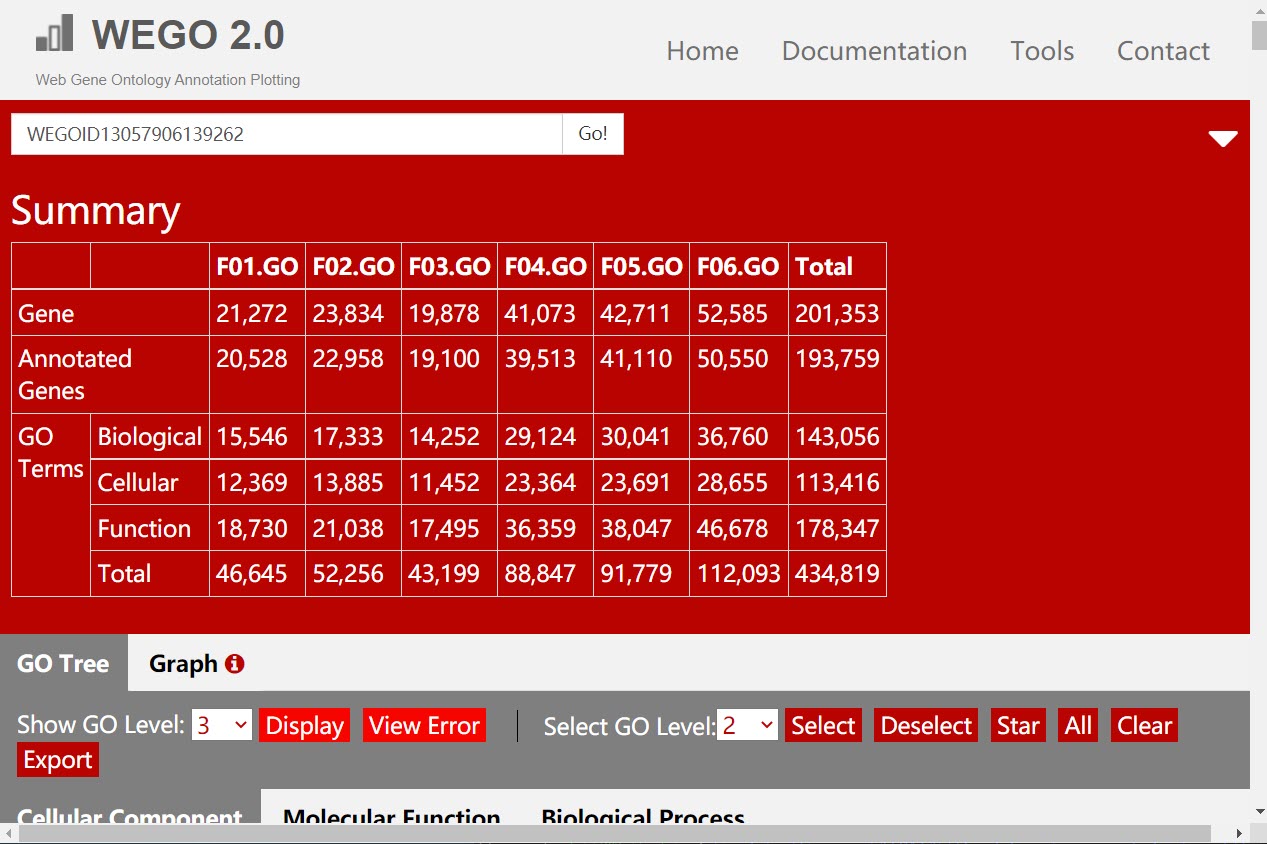
访问网页 WEGO 2.0,在网页中间位置是数据传输接口,将刚刚得到的所有结果文件拖拽上传,File format 选择Native Format,如果自己的数据是模式物种,可以在 Reference 中选择对应的物种,点击 Submit 即可。
# 脚本获取
关注公众号 “生信之巅”,聊天窗口回复 “e922” 获取下载链接。
 |  |
敬告:使用文中脚本请引用本文网址,请尊重本人的劳动成果,谢谢!
